We recently produced a set of whole-genome bisulfite sequencing experiments focused on understanding the differences in epigenetic regulation of the placenta across three closely related mouse species [1]. This data has been added to our public repository of analyzed methylomes, MethBase [2], but has some special considerations outside of the typical methylome processing that goes on for MethBase. This page is set up to explain the extra work and how the analyzed data on MethBase differs from that in the paper.
Analyzing data from closely related mouse species
While all analysis done in our study [1] was performed on raw reads mapped to the mm10 reference genome, recent publications have shown that divergence in sequence between mouse strains can have substantial effects on the observed methylation level, most often resulting in directional loss of methylation caused by C-to-T mutations in the strain being interpreted as hypomethylation during mapping to a distant reference [3].
A few months after our publication, UCSC and the Mouse Genomes project jointly developed a trackhub including a multiple alignment of several mouse strains, including those used in our paper. To improve future analysis of our data and mitigate any strain bias in methylation, we remapped each sample from our publication to its closest reference according to this trackhub. For some samples (specifically the intraspecies dataset, which was C57BL/6J) mm10 was the closest reference. For the interspecific dataset, we mapped M. m. musculus samples to PWK, M. m. domesticus to WSB, and M. spretus to SPRET.
Mapping samples to their closest references improved the mappability and eliminated the observed bias in methylation levels (comparison below). Following mapping, we used the bigMAF multiple alignment file provided by the trackhub to create one-to-one CpG liftover indices from the new reference genomes to mm10 and lifted over the methylation information using methpipe’s fast-liftover tool.
Comparison of Mapping Rate and Global Methylation Levels
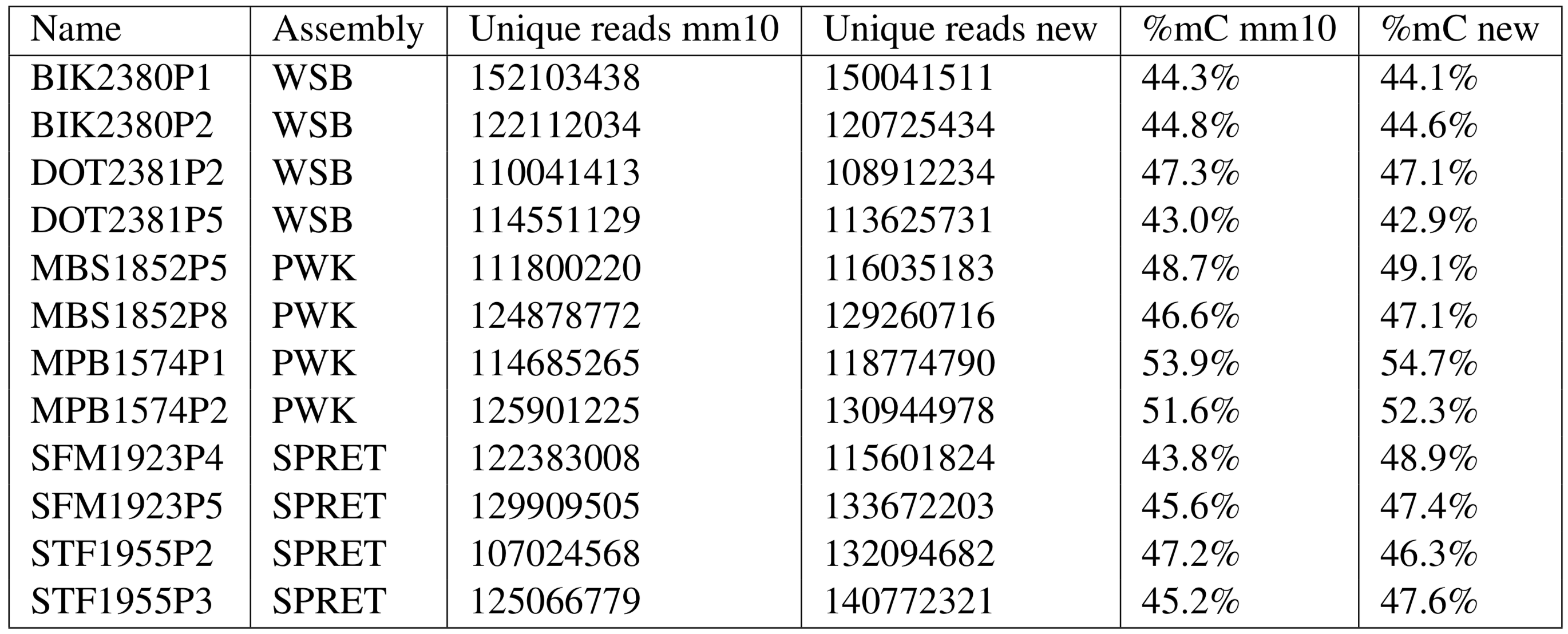
Three of four spretus samples had higher mapping rates using the spretus genome instead of mm10. Observed global methylation levels in musculus and spretus generally increased, in one case by 5.1%.
Contact
If there are any questions, suggestions, or comments about how to best access or interpret the analyzed data presented in our study, please don’t hesitate to reach out to me (Ben) at decato@usc.edu.
References
[1] Decato BE, Lopez-Tello J, Sferruzzi-Perri AN, Smith AD and Dean MD (2017)
DNA Methylation Divergence and Tissue Specialization in the Developing Mouse Placenta.
Molecular Biology & Evolution, 34(7):1702-1712 [PDF][Publisher Site]
[2] Song Q, Decato B, Hong E, Zhou M, Fang F, Qu J, Garvin T, Kessler M, Zhou J, Smith AD (2013) A reference methylome database and analysis pipeline to facilitate integrative and comparative epigenomics. PLOS ONE 8(12): e81148 [PDF] [Publisher Site]
[3] Wulfridge P, Langmead B, Feinberg AP, Hansen K (2016) Choice of reference genome can introduce massive bias in bisulfite sequencing data. bioRxiv [Publisher Site]
[4] Hickey G, Paten B, Earl D, Zerbino D, Haussler D (2013) HAL: a hierarchical format for storing and analyzing multiple genome alignments Bioinformatics, Volume 29, Issue 10, 15 May 2013, Pages 1341–1342, [Publisher Site]